Evolution of high-molecular-mass hyaluronic acid is associated with subterranean lifestyle
本次阅读的是“高分子量透明质酸的进化与地下生活方式有关”这篇论文,一作是赵阳老师!(鼓掌)
Abstract
Hyaluronic acid is a major component of extracellular matrix which plays an important role in development, cellular response to injury and inflammation, cell migration, and cancer. The naked mole-rat (Heterocephalus glaber) contains abundant high-molecular-mass hyaluronic acid in its tissues, which contributes to this species’ cancer resistance and possibly to its longevity. Here we report that abundant high-molecular-mass hyaluronic acid is found in a wide range of subterranean mammalian species, but not in phylogenetically related aboveground species. These subterranean mammalian species accumulate abundant high-molecular-mass hyaluronic acid by regulating the expression of genes involved in hyaluronic acid degradation and synthesis and contain unique mutations in these genes. The abundant high-molecular-mass hyaluronic acid may benefit the adaptation to subterranean environment by increasing skin elasticity and protecting from oxidative stress due to hypoxic conditions. Our work suggests that high-molecular-mass hyaluronic acid has evolved with subterranean lifestyle.
论文导览
介绍
裸鼹鼠 (NMR) 和盲鼹鼠 (BMR) 作为两种典型的地下动物,有长寿、强抗癌的特点,并且在两个物种的细胞外基质中均发现丰富的高分子透明质酸 (high-molecular-mass hyaluronic acid HMM-HA) 的存在。尽管部分研究阐述了两个物种之间的基因相似性与其适应底下生活的基因机制,但是 HMM-HA 在其中的作用机制仍然不明确。
Note
透明质酸 (HA) 是一种由基本二糖(D-葡萄糖醛酸及N-乙酰葡糖胺)聚合而成的糖胺聚糖,是细胞外基质的主要成分,广泛分布于结缔组织、上皮组织和神经组织当中。一般具有较大的分子质量,相对分子质量可达\(10^6\)数量级。
与多数糖胺聚糖不同,透明质酸并不是在高尔基体中被合成的,其是在细胞膜上由三种透明质酸合酶 (hyaluronan synthases HASs) ,即 HAS1、HAS2、HAS3,其作为跨膜跨糖基转移酶,固定在膜上,其催化位点嵌入到跨膜通道中,通过巧妙的双结合结构域交替向链上添加两种单体。其中 HAS2 合成分子量较大的透明质酸,已证明其在生物体中的重要地位——敲除 HAS2 基因的小鼠在妊娠期中死亡,而 HAS1、HAS3 则合成较短的透明质酸。
根据已有研究表明,小HA聚合物 (LMM-HA) 与炎症、血管生成、细胞迁移和痛觉过敏有关,HMM-HA 则与抗炎、抗痛觉过敏、抗增殖和抗应激作用有关。特别地,NMR体内发现的极高分子量HA (vHMM-HA) 已被证明具有优于人类 HMM-HA 的抗氧化应激保护。
本篇报告指出了在多种地下生物的心脏,皮肤和肾脏中均发现了丰富的透明质酸,而其在系统发育树上的相近物种中含量较少。此外,底下物种的 HA 分子量显著大于地上物种。在地下物种中 HAS2、HYAL1、HYAL2 的差异表达以及 HYAL2 上的五个独特突变帮助其积累了丰富的 HMM-HA,本研究证明丰富的 HMM-HA 已经进化为对地下环境的适应。
Fibroblasts of subterranean species secrete HMM-HA
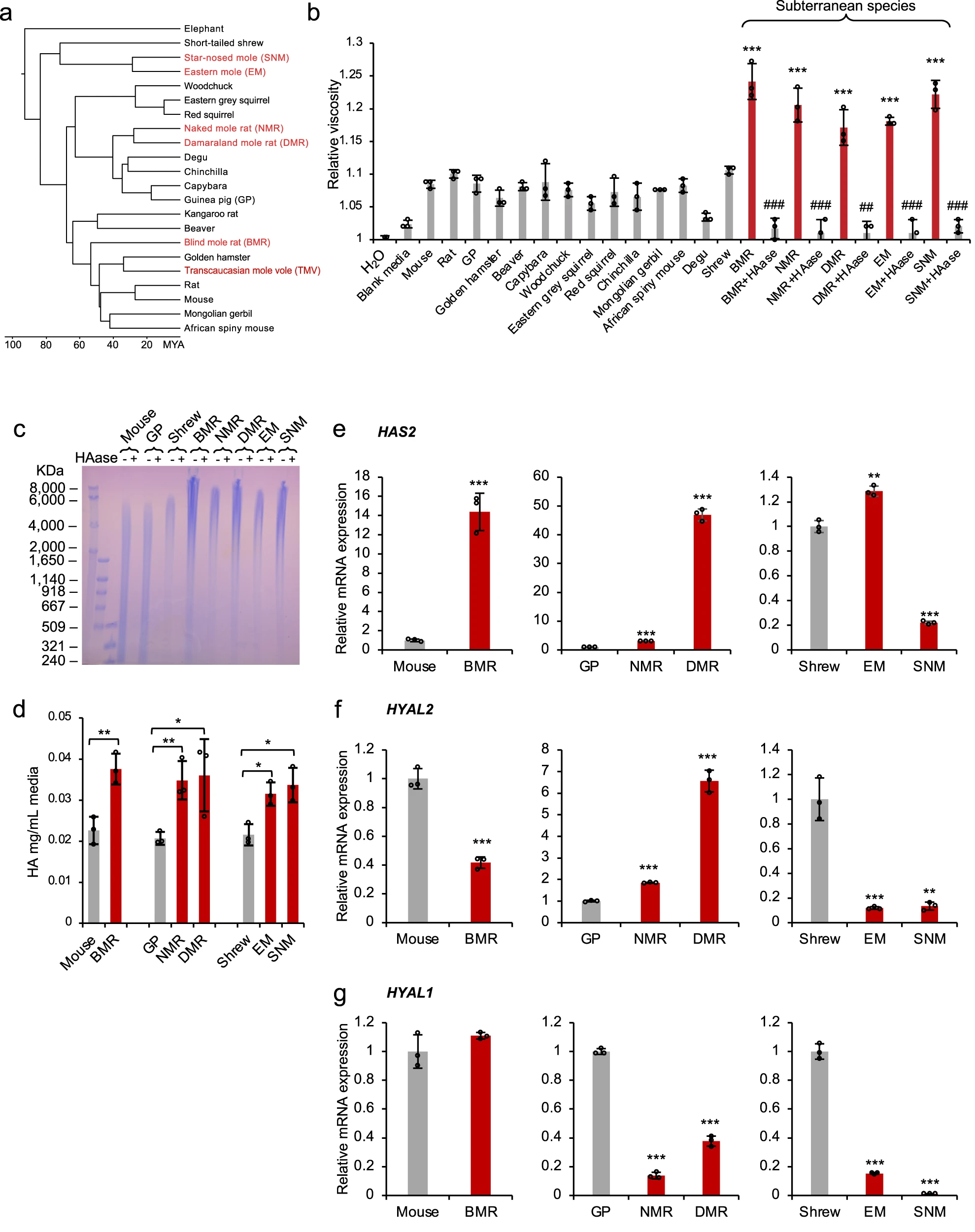
为研究丰富的 HMM-HA 是否是对地下生活的适应, 换句话说, 是否地下生物普遍具有丰富的 HMM-HA。研究员选取了六种地下物种——盲鼹鼠 (BMR, Nannospalax galili) 、裸鼹鼠 (NMR, Heterocephalus glaber) 、达马拉兰鼹鼠 (DMR, Fukomys damarensis) 、东方鼹鼠 (EM, Scalopus aquaticus) 、星鼻鼹鼠 (SNM, Condylura cristata) 和外高加索鼹鼠 (TMV, Ellobius lutescens) ,并且选取了相应的地上物种作为对照组 (图 1a) 。
研究员从这些物种中培养原代皮肤成纤维细胞 (primary skin fibroblasts) ,待其细胞生长汇合,更换培养基再培养十天后取其条件培养基测粘度。实验结果证明,除了 TMV 没有培养出细胞系以外,剩下所有地下物种的条件培养基的粘度都要显著高于地上物种。并且使用透明质酸酶 (HAase) 处理后粘度大幅降低,证明地下物种条件培养基粘度显著大于地上物种是 HA 分泌的结果 (图 1b) 。
Tip
取 3mL 蒸馏水、未使用过的 EMEM 培养基 (Blank Media) 和来自不同物种的细胞系的条件培养基在 22℃ 下通过 0.6mm Ostwald 粘度计(一种U型粘度计),不同培养基的粘度取决于其通过时间相对于蒸馏水通过时间的大小。
对于透明质酸酶处理,采用来自透明质酸链霉菌 (Streptomyces hyalurolyticus ) 的 HAase,浓度为 1U/mL,在 37℃ 下孵育过夜。
Note
这部分实验充分体现了空白对照组,对照组和实验组该如何设置。地下物种为实验组,地上物种为对照组,蒸馏水和空白培养基为空白对照组,同时对地下物种使用 HAase 处理使得实验组内形成自身对照,逐步证明了地下物种的细胞外基质含有丰富透明质酸的结论。
为直接比较细胞分泌 HA 分子量的大小,从条件培养基中纯化 HA,并通过脉冲场凝胶电泳进行分析,研究员选择了以下三个物种作为对照:
- 家鼠作为盲鼹鼠的对照。
- 豚鼠作为裸鼹鼠和达马拉兰鼹鼠的对照。
- 短尾鼩作为东方鼹鼠和星鼻鼹鼠的对照。
以上三者的选取原则都是系统发育关系较近。
实验测定HA分子量大小的方式是琼脂糖凝胶电泳和脉冲场凝胶电泳,后者相比前者可以分离更大分子量的生物大分子。电泳显示,所有 5 种地下物种的 HA 都具有高达 8000 kDa 及以上的高分子量,而 3 种地上对照的 HA 低于 6000 kDa(图 1c)。
!!! tip 脉冲场凝胶电泳 (PFGE) 脉冲场凝胶电泳在一般琼脂糖凝胶电泳的基础上实现了可改变方向的电场,从而避免了大分子量的DNA等生物大分子在一定电场下难以分离的问题。具体原理可见参考资料4, 5.
研究员通过咔唑定量测定了培养基中HA的量,实验证明地下物种分泌的HA量均高于地上物种 (图 1d)。
Tip
透明质酸中含有等量摩尔比的 N-乙酰氨基葡萄糖和葡萄糖醛酸,用硼砂作催化剂对透明质酸钠用硫酸进行酸解,能将葡萄糖醛酸分离出来。葡萄糖醛酸与咔唑反应形成有机络合物,该络合物显示特有的紫色,其吸光度和葡萄糖醛酸的浓度成正比。通过葡萄糖醛酸的含量能确定透明质酸钠的含量。
Warning
咔唑可能与培养基中潜在的其他糖类发生反应,造成测定上不可避免的误差。实验中同样采用了 HA ELISA 方法定量测定HA。
ELISA (Enzyme-linked immunosorbent assay),即酶联免疫吸附法,利用经典的抗原-抗体特异性结合和酶显色反应来标定特定分子的存在,并可做到定量测定。更多可见参考资料7.
接着研究员对负责 HMM-HA 组织水平的三个基因的表达水平:透明质酸合酶2 (HAS2)、透明质酸酶-1 (HAYL1)、透明质酸酶-2 (HAYL2)进行了测定,结果发现了不同地下物种相对其近缘地上物种的三种基因表达有差异化取舍。但在总体上,地下物种相对于地上物种,通过差异化调节HAS2、HYAL1、HYAL2基因表达,实现了分泌更高的分子量和更多的 HA (图1e-g).
Subterranean species have higher HA abundance in tissues due to differential expression of HA synthases and hyaluronidases
研究员率先比较了体内各组织透明质酸含量的多少,在人体中,皮肤拥有全身50%的透明质酸,而在啮齿类动物体内,心脏和肾脏也含有较多透明质酸。于是研究员取实验动物皮肤、心脏和肾脏的冷冻切片,用荧光标记的透明质酸结合蛋白 (HABP) 染色,通过倒置荧光显微镜定量测定,染色结果显示所有六种地下物种在皮肤中的 HA 含量都明显高于其地上对照 (图2a) 。采用 HAase 处理后均为阴性染色。以上结果表明,地下物种在其组织中产生更大量的 HA。
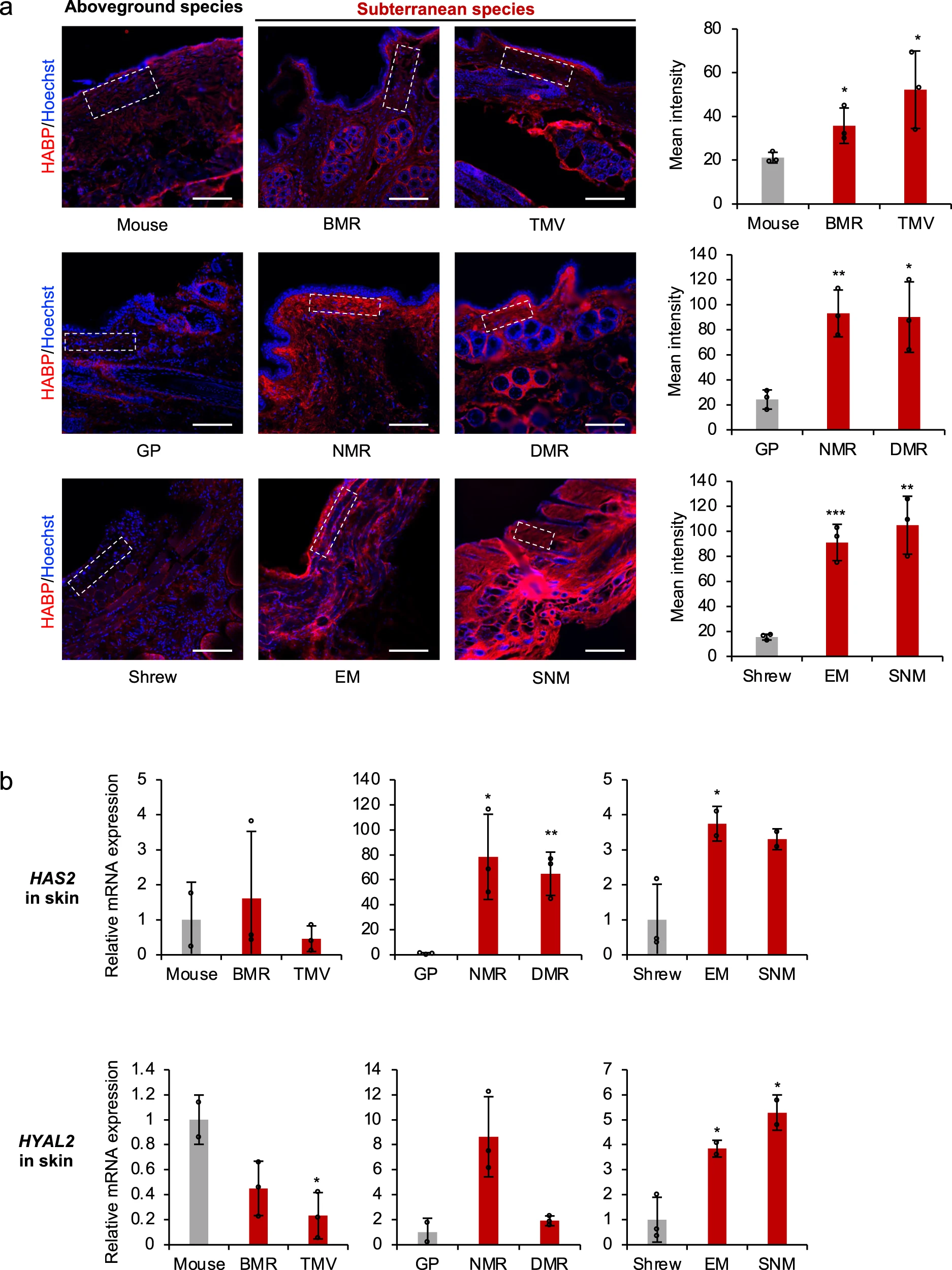
接下来研究员使用 RT-qPCR (按照wiki的约定,应该是qRT-PCR,定量逆转录聚合酶链式反应)检测了 HAS2、HYAL2、HYAL1 的表达水平,发现 BMR 和 TMV 的 HAYL2 表达水平显著下降,NMR 和 DMR 的 HAS2 表达水平显著上升, EM 和 SNM 的 HAS2 和 HAYL2 的表达水平都有显著提高,推测存在其他调节因子促使 EM 和 SNM 在皮肤中拥有高丰度的 HA。这些结果表明,地下物种差异调节其皮肤中 HAS2 和 HYAL2 的表达,以产生更丰富的 HA。
Tip
这个技术基于一般的PCR改进而来,特点就包含在名字里:定量和逆转录,其中定量PCR (qPCR) 可以定量测定模板含量,一般采用荧光信号作为定量测量的信号。
qPCR对基因表达的定量方法与数学证明请参见相关证明。
研究员对心脏和肾脏进行了同样的实验,发现三种基因在心脏与肾脏中的调节方式和皮肤有所区别,这表明尽管 HA 在地下物种的不同组织中积累,但它们的调节机制是组织特异性的。
Evolution of HA degrading genes in subterranean species
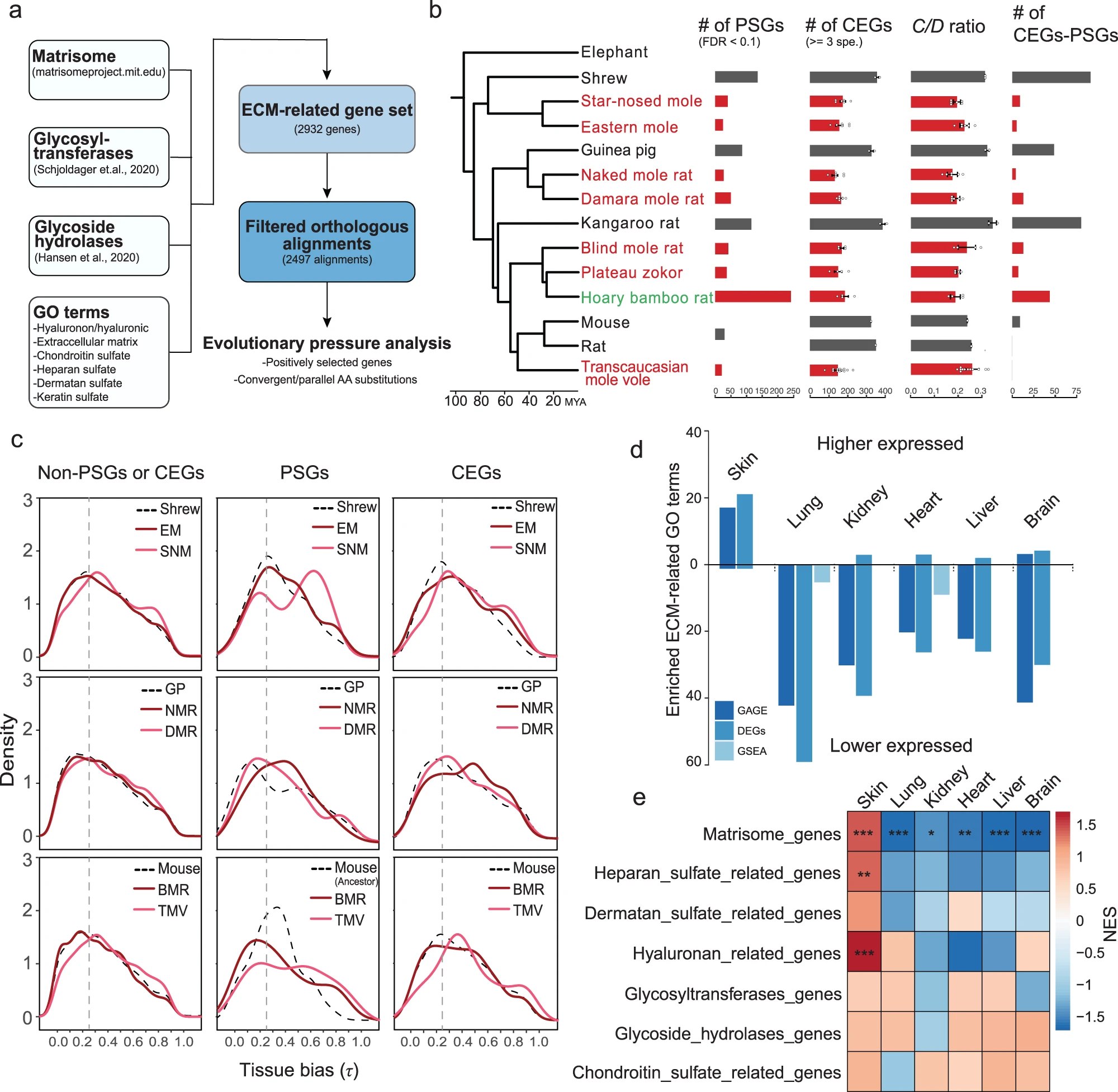
就前文所提及的研究结果而言,地下物种体内 HA 的丰度因组织而异。所以影响样本的因素包含有物种与组织两个维度,为了系统评估 HMM-HA 在不同组织中的调节,研究员决定通过 RNA-seq 定量测量地上物种与地下物种的 6 个组织(皮肤、大脑、肺、心脏、肾脏和肝脏)的样本。通过对所有物种的8417个同源基因的总体表达相似性矩阵的分层聚类表明,样本主要按照组织而非物种分类 (补充图S6a)。
基于以上结论,研究员对来自同一组织的样本的表达数据进行分组、过滤和归一化,归一化结果显示每组内中位表达值相似 (补充图S6b)。
Normalization——归一化
在生物信息学分析中,归一化是一种很重要的数据处理手段,它可以有效消除批次效应带来的意外差异,当然这么说十分抽象,让我们从实际出发:本篇论文中基于 DESeq2 进行归一化处理,同样还有 TMM 归一化方法,两者所面临的问题都是:我们不能保证所有批次样品之间的无关变量都是控制良好的。
比如湿度温度,机器的状态,送检样本总量,这些都会导致我们最终的测序结果不同。最简单的事实就是,假如我第二次提取出 RNA 的手艺进步了,损失少了一点,那送检 RNA 总量就比第一次多,那我最终测出的“第二次样品中某个基因表达相比第一次偏高”的结论就是不靠谱的。更糟糕的是,我们甚至不知道有什么无关变量无意间被改变了。
基于对这个问题的察觉,我们向 RNAseq 引入归一化方法:DESeq2 和 TMM,两者建立在同一个基本认识上:大多数基因的表达都是非差异化的,比如很多管家基因在不同细胞内的表达程度是相近的。这点共识可以推导出如下结论:我们很显然可以在多组数据的对比之下找出那些不被差异化表达的基因,在我们尽可能控制无关变量的前提下,这些不差异化表达的基因的测序结果差异通常呈现稳定的倍数关系(比如移液枪多吸了一点点)。
那么我们通过一个共识的“基准”,就可以衡量所有样本在测序上“吃的亏”或者“不当获利”,从而通过适当的修正,使得所有样本能在同一个假定的公平起点上比较。
无论是 DESeq2 还是 TMM 都会率先根据总体样本计算出“缩放因子”,通过对每个样本以特定的缩放因子进行修正,来修正由于各种可能导致的原始文库大小差异、机器过程差异甚至是不为人知的玄学差异。从而使得我们最终的结果具有可比性。
Note
踩坑记录:AI中常用的归一化方法和标准化方法和生信中的归一化方法不是一回事。前者归一化是简单的范围缩放为 [-1, 1],标准化则是使原始分布转化为期望为0,方差为1的标准分布。后者归一化是广义上的归一化,虽然沿用了 normalization ,但是实际算法要复杂的多,是一种基于整体样本分布进行一般化调整的算法。
研究员首先从 RNAseq 数据中检测了 HAS2、HYAL1 和 HYAL2 的表达,这与 qPCR 结果基本一致 (补充图S5b),这再次印证了前文所提的地下物种差异调节其皮肤中 HAS2 和 HYAL2 的表达,以产生更丰富的 HA论点。
基于RNA-seq的结果 (图3b),研究员对更多基因的表达情况进行了探究。HA 由三种 HA 合酶 (HAS1-3) 合成,并被 6 种透明质酸酶 (HYAL1-6) 和两种新发现的 HA 降解酶 CEMIP (KIAA1199)26 和 CEMIP2 (TMEM2) 降解,并且 HA 的代谢还与相应 UDP 糖代谢状况相关,因此研究了参与 UDP-GlcNAc 合成、UDP-GlcUA 合成、透明质酸合成、透明质酸降解、透明质酸结合和 TNFAIPs 的基因的表达变化。
研究员对此提出如下观察结果:大量 HA 结合基因在地下物种中表达更高;HA 降解基因在地下物种的多个组织中表达较低 。对此提出 HA 降解途径的缺陷可能导致 HA 在地下物种中积累高数量和高分子量。
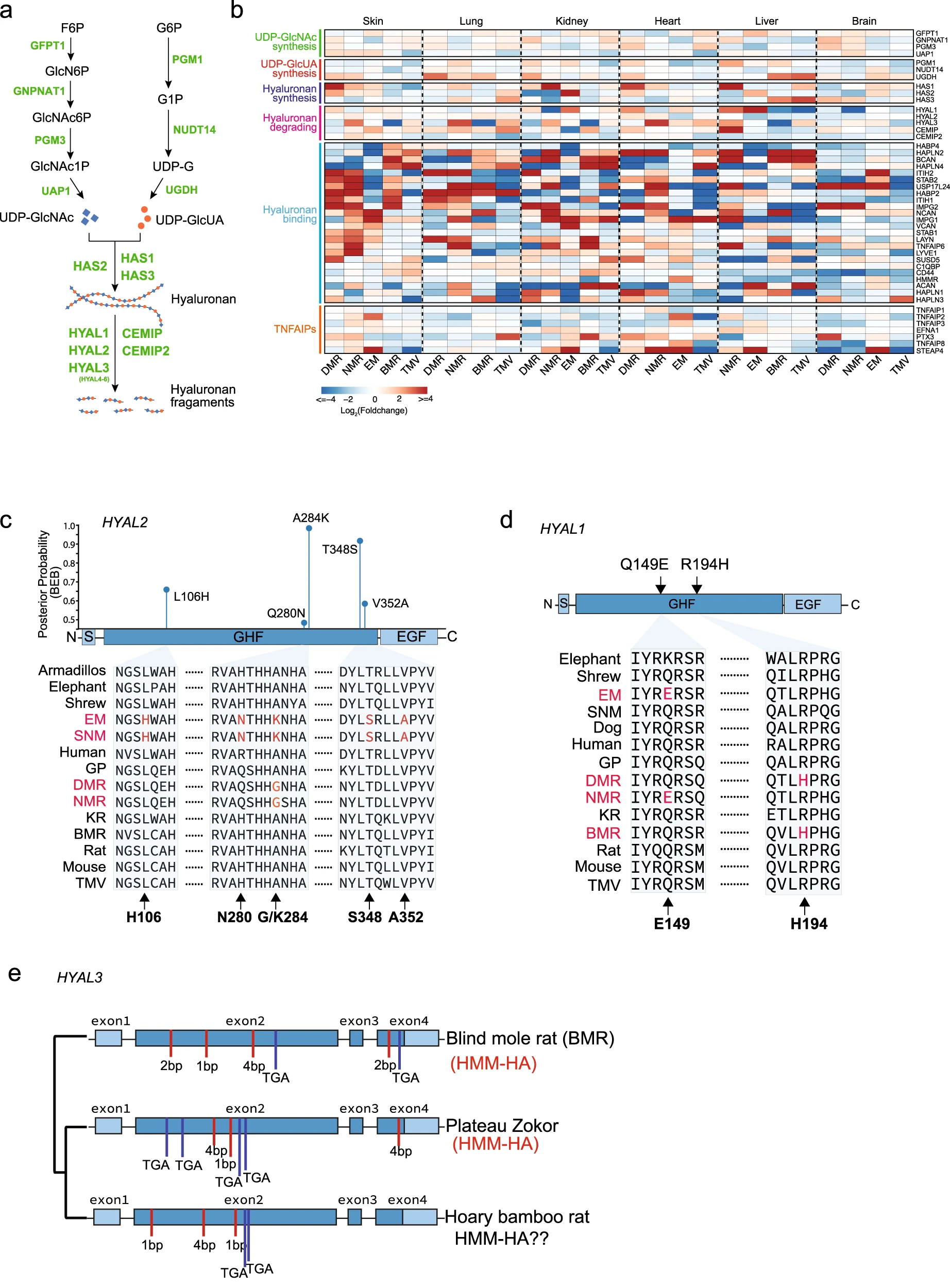
基于地下物种的 HA 降解途径缺陷对 HA 的积累可能存在贡献的结论,研究人员进一步探究参与 HA 降解的基因的进化过程。其中采用了分支位点模型和贝叶斯经验贝叶斯 (BEB) 的数学方法,筛选出了 SME 和 EM 共同祖先在 HYAL2 上的正向选择位点,并且证明其在关乎水解酶活性的 GHF 结构域中。在其他 HA 分解酶上也有相似的修饰 (图3c、3d)。
同义替换与非同义替换——进化之择
我们都知道的是,一段基因越是保守,对该生物就越是重要,也意味着这段基因足以应对当前的生存压力,冒然改变带来的风险远大于收益。
而在基因变异当中,碱基替换存在同义替换与非同义替换两种类型(密码子简并),同义替换对蛋白质结构无影响,而非同义替换严重可致死,当我们不考虑变异结果时候,可认为同义替换和非同义替换是等概率的。假如我们记前者为\({\rm dS}\),后者为\({\rm dN}\),记\(\omega={\rm dN/dS}\),则我们有以下三种情况:
- \(\omega=1\):表示中性进化,意味着两者发生频率相同,意味着自然选择压力对这种基因的变异不太关心。
- \(\omega < 1\):代表纯化选择或负向选择,意味着非同义替换大概率有害,会被自然选择清除。
- \(\omega > 1\):代表适应性进化或正向选择,表明在当前的选择压力下非同义替换大多起到了积极性作用,提高了物种存活率。
于是我们通过简单的比值量化了自然选择,这将是我们分析基因变异贡献的重要理论基础。
分支位点模型
分支位点模型基于正向选择通常发生在少数几个物种分支上,并广泛影响其后代的思想,其的模型基于进化树结构,提出了前景分支和背景分支的思想:
- 前景分支:可能发生正向选择的进化分支。
- 背景分支:其余非前景分支的进化分支。
模型通过似然比检验法验证假设,通过比较前景分支和背景分支的基因差异,给出前景分支上是否可能存在正向选择的位点的建议。
本篇论文采用了 PAML 4 中的 Model A,可见参考资料12.
贝叶斯经验贝叶斯 (BEB) —— 用数据说话
BEB 基于贝叶斯估计和经验贝叶斯,传承了贝叶斯估计的基本思想:先验分布产生数据集,数据集修正先验分布,得到可信的后验分布。
经验贝叶斯作为贝叶斯估计的一种改进,拒绝主观定义先验分布,而是先从数据集中猜出一个决定先验参数分布的超参数,然后得出我们需要的先验分布。
至于 BEB 则改进了小样本量下 EB 过度自信的缺点,通过继续估计超参数的超分布,在考虑其置信区间的前提下给出后验分布,比较适合生物学这种小样本的场景。
在本篇论文中,已知实验的模型参数和和给定实验数据,BEB会给我们计算一个后验概率 PP 表明特定位点的突变是正向选择的概率有多少,如果后验概率超过设定的阈值则认为该基因突变是正向选择的结果。
据以往研究,BMR 和 NMR 中的基因丢失有助于适应地下环境,因此研究者进一步研究了地下生物中 HA 相关基因的假基因化,发现 BMR 中 HYAL3 出现假基因化(移码突变和过早终止密码子),有显著松弛选择性 (图3e) 。总的来说,这些研究结果暗示透明质酸酶基因在地下物种中经常经历选择压力。
假基因化
假基因是一类染色体上的基因片段,其序列通常与对应的正常基因类似,但几个关键位点发生了突变,使其无法表达蛋白质或表达产物无功能。
假基因化描述的是正常基因变为假基因的过程。
RELAX与松弛选择性
没研究,参见参考资料14.
Differential expression and sequence changes in HA synthases and hyaluronidases contribute to HMM-HA in subterranean species
为了从机制上评估 HA 相关酶的差异化表达和序列变化对地下物种中 HMM-HA 积累的贡献,研究员在 293T 和 Hela 细胞中过表达了来自不同物种的 HAS2、HYAL2 或 HYAL1。其中分别来自小鼠和BMR的 HAS2 编码质粒以1:14比例转染细胞,以模拟两者不同的表达量,采用 RT-qPCR 检测表达量 (图S7)。转染后两天从条件培养基中纯化 HA 进行电泳,结果证明转染了 BMR HAS2 的细胞所表达的 HA 无论是数量还是分子量都大于转染小鼠 HAS2 的细胞 (图S8a,图4a),有趣的是,即使两者表达水平相同(转入相同数量的质粒),表达 BMR HAS2 的细胞分泌的 HA 的分子量还是更大。特别地,各方面表现都比小鼠 HAS2 要好的 BMR HAS2 和前者只有四个氨基酸的差别。因此,这一结果表明,BMR HAS2 中的四个突变,结合其较高的表达,有助于 BMR 中 HMM-HA 的积累。同样,表达 SNM HAS2 的细胞也比表达 shrew HAS2 的细胞产生更大大小的 HA (图4b)。这些结果表明,HAS2 的序列变化和差异表达水平有助于 HMM-HA 在地下物种中的积累。
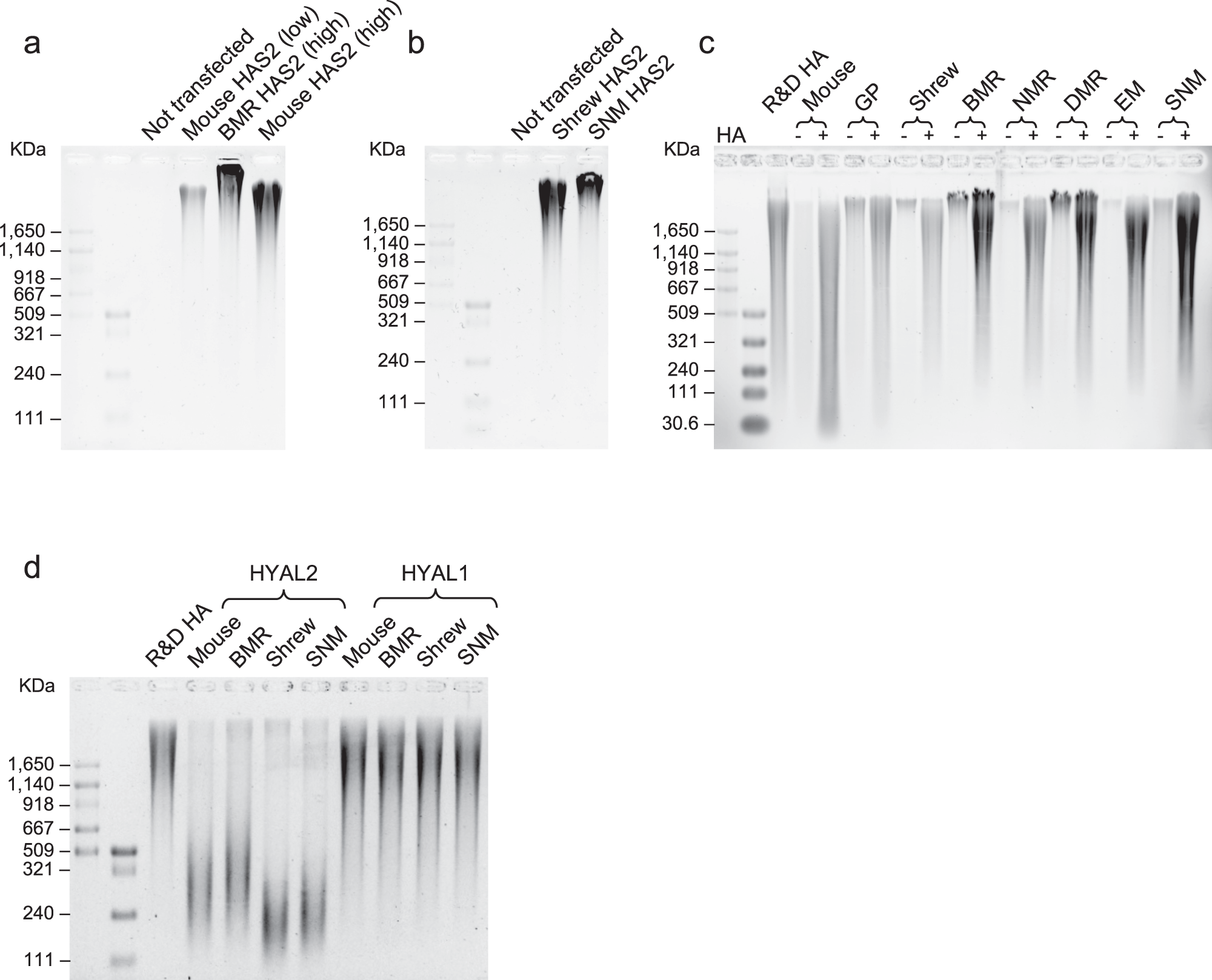
接下来研究员比较地下与地上物种之间的 HA 降解。取商用 HMM-HA 与5种地下生物或3种地上对照生物的已汇合成纤维细胞孵育两天,实验结果是地下物种细胞的培养基中残留 HA 的量多于地上物种。这些结果在生长期细胞中得到了复现,证明地下物种的细胞对 HA 的降解较少。
有了这个实验基础,研究员进一步验证来自不同物种的 HA 降解酶 HYAL1 和 HYAL2 在活性上的差异。在 293T 中过表达小鼠、BMR、鼩鼱或 SNM 的 HYAL2 或 HYAL1。将商业 HA 与转染细胞一起孵育 2 天,提取并进行电泳。实验结果中鼩鼱 HYAL2 比 SNM HYAL2 诱导了更多的商业 HA 降解 (图S8c, d),表明地下物种中的 HYAL2 具有较弱的 HA 降解活性。值得强调的是,小鼠的 HYAL2 降解了更多的 HA,也导致剩余 HA 的分子量要小于 BMR 和 SNM (图4d)。而表达不同物种的 HYAL1 的细胞在 HA 降解方面没有显示出显着差异 (图4d、图S8e),研究员推测因为 HYAL1 是一种细胞内酶。综上所述,这些结果表明 HYAL2 在导致 HMM-HA 积累的地下物种中的活性较低。
细胞内酶
由细胞合成的,在细胞内发挥生物学作用的酶类,与之相对的是细胞外酶,后者被分泌到细胞外发挥作用。
Structural influences of EM and SNM HYAL2 mutations
EM 和 SNM 的 HYAL2在结构上鉴定出五个氨基酸的不同,研究人员为推断其在地下生物积累 HMM-HA 中所起的作用,将其蛋白质结构与人 HYAL2 作对比,初步锁定了各个残基的作用。
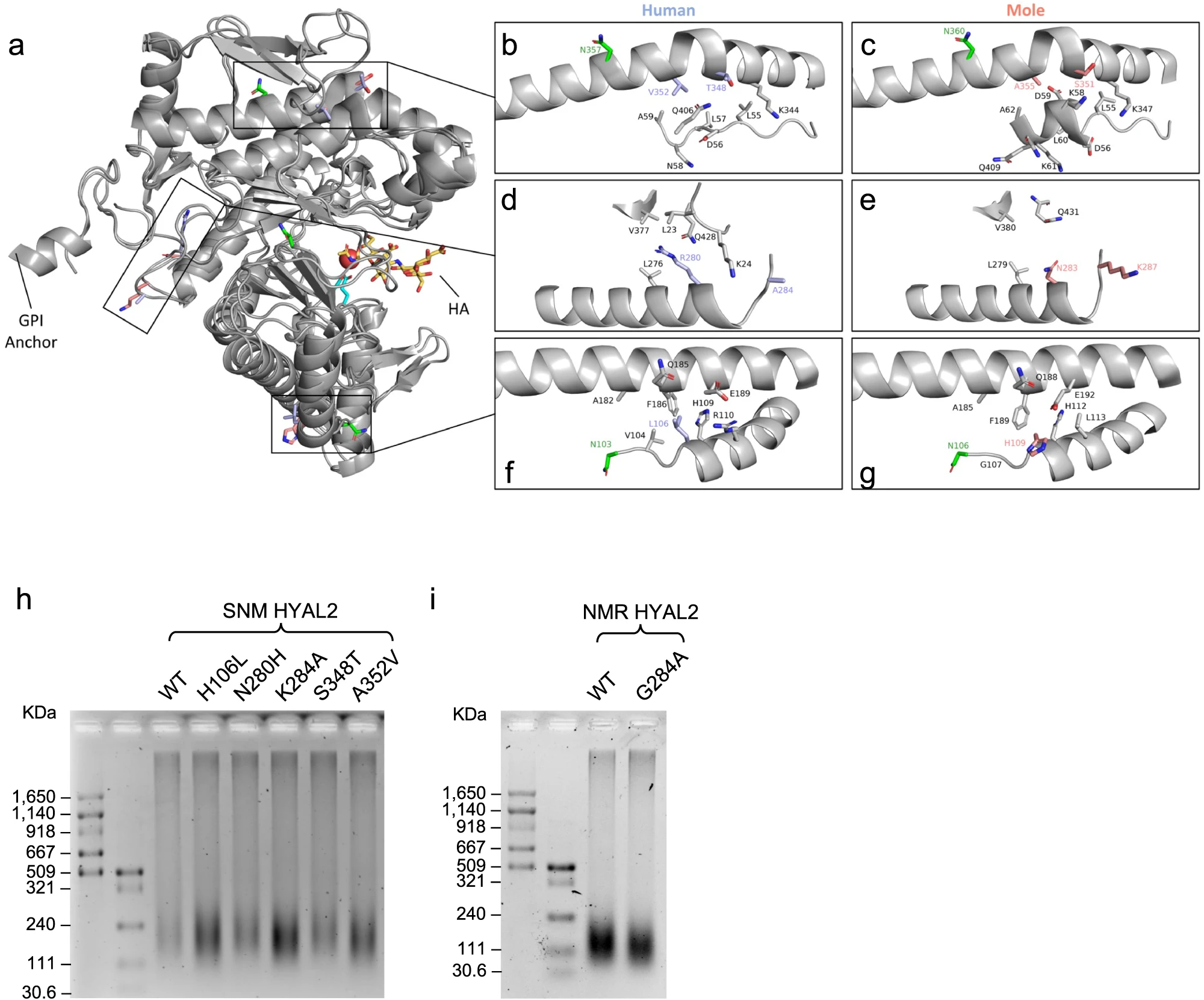
之后基于对五个位点定向突变为地上生物氨基酸,对各个 HYAL2 进行酶活性检测,发现五个残基的突变都能有效提高 HYAL2 的活性。总的来说,在 EM 和 SNM 中观察到的突变可能会影响 HYAL2 的蛋白质稳定性和翻译后修饰。
Note
这部分没怎么看懂。
HMM-HA coevolves with ECM in subterranean species
鉴于 HA 是细胞外基质 (ECM) 的主要成分之一,研究人员推测地下生物演化为 HMM-HA 丰富表型的过程必然和 ECM 的变化有密不可分的关系。
更多
DESeq2 和 TMM 算法理解
准确来说,DESeq2 是一个功能强大且完善的统计包,在这篇文章中用到的部分主要是其的比率中位数方法。有如下数学定义:
假设基因为i,样本为j,那么我们规定\(K_{ij}\)为“在样本j中的基因i的RNA-seq计数值”,假设总样本数量为m,对于每个样本j,我们有
我们称\(s_j\)为归一化常数,对于每个基因i均有一个归一化常数\(s_{ij}\),我们规定\(s_{ij}=s_j\).
上面这个式子看着有点绕,实际上用语言表述出来就是:取所有样本中同一基因的计数值的几何平均值作为分母,取特定样本中的该基因作为分母,求得一系列比率,然后在同一组中取所有基因各自比率的中位数作为本组的归一化常数。最终的归一化采用本组内各基因计数除以本组归一化常数得到归一化结果。
这种处理方法是很高明的,首先是对不同组的同一基因分别求几何平均值和相对其几何平均值的比值。几何平均值对极端值不敏感,意味着即使组别效应影响了几组基因,几何平均值也能较好反应出一般情况下的虚拟参考值。而各样本组内基因与其的比值则反应了不同组之间同一个基因相对表达水平的高低。
一旦我们对所有组里的所有基因都做完这个比率计算后,我们就在每一个样本里得到了所有基因在总体样本下计算出的比率信息,这里选择了所有比率的中位数是有深意的,如果组别效应足够明显,会导致特定组内特定基因表达的极高上调或者下调,而真正稳定表达的基因往往是多数,通常会占据数据的中间部位,如果我们使用中位数,就能获取到它们的比率。而稳定表达的基因在不同样本之间的差异往往是测序深度引起的,比率能更好反应其测序深度差异,从而在最终的归一化中排除测序深度差异的问题。
TMM 则采用了一种更加复杂的方法来估计归一化常数,但是其和DESeq2的比率中位数方法没有本质的差别,实际上都是考虑测序深度与文库总量的差异对测序结果的影响,并尝试消除它。
TMM 定义\(Y_{gk}\)为原始数据中,文库k中基因g的计数值,\(\mu_{gk}\)定义为文库k中基因g未知的真实表达水平(转录本数),\(L_g\)定义为基因g的长度,\(N_k\)定义为文库k的总reads数,显然\(Y_{gk}\)的期望可表达为
其中\(S_k\)代表样品的总RNA输出量。
显然这个期望很受文库大小的影响,除了基因自身表达水平外,主要取决于总RNA输出和文库总reads数,而后两者就是我们需要解决的文库大小与测序深度问题的关键所在。
\(N_k\)是一个很容易知道的量,但是\(S_k\)则难以估计,因为我们无法准确知道所有基因的真实表达水平与准确长度,但是对于大多数非差异性表达的基因来说,其真实表达水平近乎视作常量,显然对于同一个基因来说,其准确长度也是相同的。基于大多数基因稳定表达的假设,我们可以认为不同样品间的总RNA输出量比值\(f_k=S_k/S_{k'}\)等价于文库大小的倍数变化。那我们的目标转变为寻找那些稳定表达的基因,这和DESeq2的思路是类似的。
在正式开始计算前,TMM 需要确定一个参考样本,参考样本的选取标准通常是对照组或中位文库大小的组,其他组均以参照组为标准进行计算。TMM 定义两个统计量,第一个是样本间对数变化特征\(M_g\),定义为
第二个是绝对表达水平\(A_g\),定义为
上两式中文库\(k'\)就是参照组。
依据两个统计量对原始数据进行修剪,一般修剪M值上下共30%和A值上下共5%,此步骤称为截尾修剪,目的是去除异常表达的基因。两个步骤一般并行进行。特别地,0值不会参与到最后的运算中。
对于修整过的数据集,我们计算截尾加权对数均值\(log_2(TMM_k^{(r)})\),其定义为使用参考样本r的样本k的归一化常数的对数
特别地,\(Y_{gk}, Y_{gr}>0\).
需要强调,任意计数为0的在计算前都应该被修剪掉,而\(G^*\)代表未经修剪切具有有效M值和A值的基因集。
而将结果取指数则得到了对应的归一化常数TMM。
显而易见的是 TMM 也是围绕找到稳定表达的基因并且借此判断测序深度影响这个核心思路来对数据进行归一化处理的。特别地是其在求归一化常数的过程中进行加权平均,权重是M值对应近似方差的倒数,这意味着越稳定的基因权重越高,从而更能准确定位出代表该样本的测序深度影响因子,也就是归一化常数。
Reference
- 玻尿酸 - 维基百科,自由的百科全书
- Hyaluronan synthase - Wikipedia
- Viscometer - Wikipedia
- Pulsed-field gel electrophoresis | Nature Protocols
- What is Pulsed Field Gel Electrophoresis? | NEB
- QB/T 4576-2023 透明质酸钠
- ELISA | 原理介紹 - ACE Biolabs
- qPCR (real-time PCR) protocol explained - YouTube
- Reverse transcription polymerase chain reaction - Wikipedia
- A scaling normalization method for differential expression analysis of RNA-seq data | Genome Biology | Full Text
- Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2 | Genome Biology | Full Text
- PAML 4: Phylogenetic Analysis by Maximum Likelihood | Molecular Biology and Evolution | Oxford Academic
- 假基因 - 维基百科,自由的百科全书
- RELAX: Detecting Relaxed Selection in a Phylogenetic Framework | Molecular Biology and Evolution | Oxford Academic